Learning Amino Acids Part 1: Non-Polar Amino Acids, Rodrigues Rotation, and Lennard-Jones Potential
By Ken Koon Wong in r R amino acid non-polar rodriguez rotation formula lennard-jone potential plotly
June 7, 2026
🧬 Back to basics! Learning non-polar amino acids, what zwitterions actually are, and dipping into the applied math — Rodrigues rotation and Lennard-Jones potential. Slowly building toward optimal phi/psi!
Motivations
We’ve explored quite a bit lately in molecular dynamic simulation and then protein-protein docking as well the last time. There is still so much to learn. I’ve decided to go back to basics, revisiting our old friends amino acids and try to understand the natural properties behind each one and see if that will make more sense in the future when we’re exploring more. While making notes for myself of all the amino acids, I’ll also try to understand some of the basic math behind the structures. Are you ready !? Lol, I’m not, but let’s go anyway! 🤣
Objectives:
- Amino Acids
- Rodriguez Rotation Formula
- Lennard-Jone Potential Enegery
- Opportunities For Improvement
- Lessons Learnt
Amino Acids
Amino acids are the building blocks of proteins, each sharing a common backbone: a central α-carbon bonded to an amino group (–NH₂), a carboxyl group (–COOH), a hydrogen atom, and a variable side chain (R group) that defines each amino acid’s identity and chemistry.
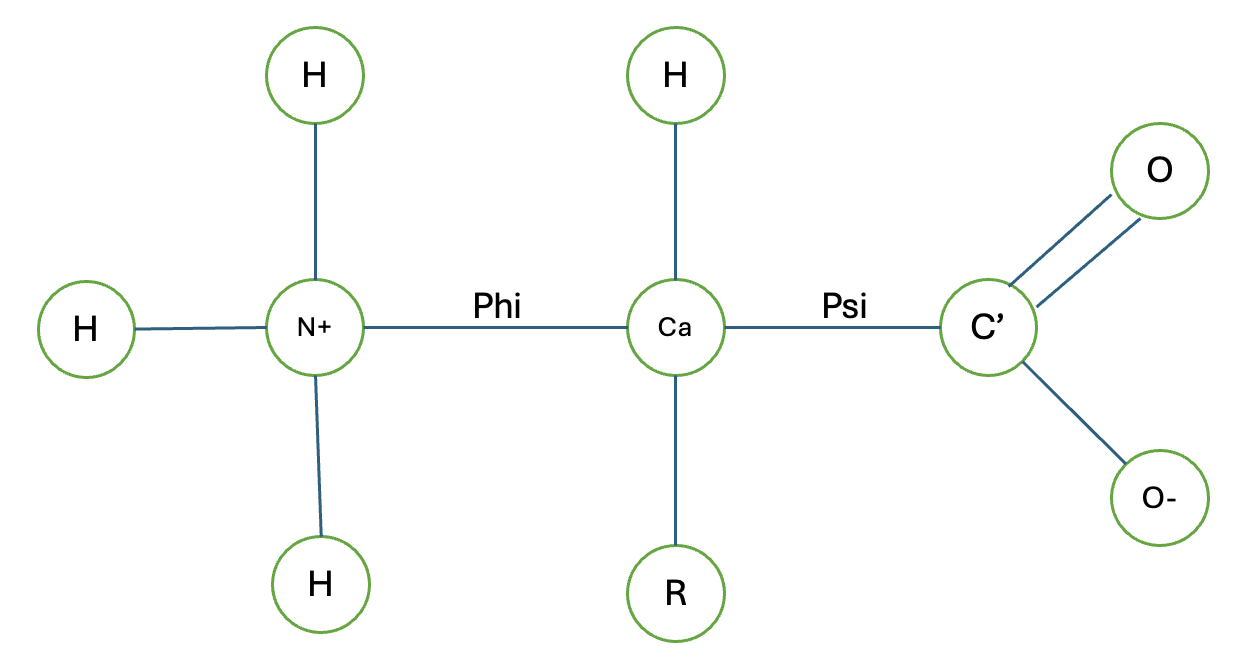
Non-polar amino acids
Non-polar amino acids have hydrophobic side chains — they avoid water and tend to cluster in the interior of folded proteins, forming the hydrophobic core that drives protein stability. Understanding each one’s shape and bulk is directly relevant to how they pack, how they constrain backbone flexibility, and how substitutions affect enzyme active sites.
library(tibble)
library(kableExtra)
aa_nonpolar <- tribble(
~aa, ~aa3, ~name, ~functional_group, ~smiles_sidechain, ~charge_ph7, ~mw_da, ~pka, ~md_note, ~main_function,
"G", "Gly", "Glycine", "H (none)", "[H]", "Neutral", 75.03, NA_real_, "Minimal VDW radius; unrestricted phi/psi; near-zero excluded volume", "Conformational flexibility; tight turns; active site geometry",
"A", "Ala", "Alanine", "Methyl", "C", "Neutral", 89.09, NA_real_, "Low steric perturbation; high alpha-helix propensity in force fields", "Helix former; hydrophobic core; alanine-scanning mutagenesis",
"V", "Val", "Valine", "Isopropyl", "CC(C)", "Neutral", 117.15, NA_real_, "Beta-branching restricts psi; favors extended beta-sheet; large gamma-carbons", "Beta-sheet core; hydrophobic packing; sickle-cell HbS Glu6Val",
"L", "Leu", "Leucine", "Isobutyl", "CCC(C)C", "Neutral", 131.17, NA_real_, "Flexible chi2; common rotamers at -65/-65 and -65/175; high hydrophobic SASA", "Hydrophobic core; leucine zippers; most abundant non-polar in proteomes",
"I", "Ile", "Isoleucine", "sec-Butyl", "CCC(C)", "Neutral", 131.17, NA_real_, "Beta-branching + gamma-branch; most restricted chi1/chi2; large buried SASA", "Hydrophobic core; beta-barrel interiors; transmembrane helices",
"P", "Pro", "Proline", "Pyrrolidine ring", "C1CCNC1", "Neutral", 115.13, NA_real_, "Fixed phi ~-60; no backbone NH donor; cis/trans isomerism at Xaa-Pro bond", "Helix breaker; beta-turns; collagen Gly-Pro-X repeats",
"F", "Phe", "Phenylalanine", "Benzyl", "Cc1ccccc1", "Neutral", 165.19, NA_real_, "Rigid aromatic ring; pi-pi stacking and cation-pi in MD energy decomposition", "Hydrophobic core; aromatic clusters; ligand binding pockets",
"W", "Trp", "Tryptophan", "Indolylmethyl", "Cc1c[nH]c2ccccc12", "Neutral", 204.23, NA_real_, "Indole NH can H-bond; amphipathic at membrane interface; strong 280nm absorbance", "Membrane anchoring; fluorescence probe; ligand binding; rarest standard AA",
"M", "Met", "Methionine", "Thioether", "CCSC", "Neutral", 149.20, NA_real_, "Flexible sulfur geometry; oxidizable to sulfoxide in long MD runs; check reactive FF", "Translation initiation; hydrophobic core; redox sensing"
)
aa_nonpolar |>
dplyr::select(aa:mw_da) |>
kbl()
|
aa |
aa3 |
name |
functional_group |
smiles_sidechain |
charge_ph7 |
mw_da |
|---|---|---|---|---|---|---|
|
G |
Gly |
Glycine |
H (none) |
[H] |
Neutral |
75.03 |
|
A |
Ala |
Alanine |
Methyl |
C |
Neutral |
89.09 |
|
V |
Val |
Valine |
Isopropyl |
CC(C) |
Neutral |
117.15 |
|
L |
Leu |
Leucine |
Isobutyl |
CCC(C)C |
Neutral |
131.17 |
|
I |
Ile |
Isoleucine |
sec-Butyl |
CCC(C) |
Neutral |
131.17 |
|
P |
Pro |
Proline |
Pyrrolidine ring |
C1CCNC1 |
Neutral |
115.13 |
|
F |
Phe |
Phenylalanine |
Benzyl |
Cc1ccccc1 |
Neutral |
165.19 |
|
W |
Trp |
Tryptophan |
Indolylmethyl |
Cc1c[nH]c2ccccc12 |
Neutral |
204.23 |
|
M |
Met |
Methionine |
Thioether |
CCSC |
Neutral |
149.20 |
aa_nonpolar |>
dplyr::select(aa,aa3,md_note,main_function) |>
kbl()
|
aa |
aa3 |
md_note |
main_function |
|---|---|---|---|
|
G |
Gly |
Minimal VDW radius; unrestricted phi/psi; near-zero excluded volume |
Conformational flexibility; tight turns; active site geometry |
|
A |
Ala |
Low steric perturbation; high alpha-helix propensity in force fields |
Helix former; hydrophobic core; alanine-scanning mutagenesis |
|
V |
Val |
Beta-branching restricts psi; favors extended beta-sheet; large gamma-carbons |
Beta-sheet core; hydrophobic packing; sickle-cell HbS Glu6Val |
|
L |
Leu |
Flexible chi2; common rotamers at -65/-65 and -65/175; high hydrophobic SASA |
Hydrophobic core; leucine zippers; most abundant non-polar in proteomes |
|
I |
Ile |
Beta-branching + gamma-branch; most restricted chi1/chi2; large buried SASA |
Hydrophobic core; beta-barrel interiors; transmembrane helices |
|
P |
Pro |
Fixed phi ~-60; no backbone NH donor; cis/trans isomerism at Xaa-Pro bond |
Helix breaker; beta-turns; collagen Gly-Pro-X repeats |
|
F |
Phe |
Rigid aromatic ring; pi-pi stacking and cation-pi in MD energy decomposition |
Hydrophobic core; aromatic clusters; ligand binding pockets |
|
W |
Trp |
Indole NH can H-bond; amphipathic at membrane interface; strong 280nm absorbance |
Membrane anchoring; fluorescence probe; ligand binding; rarest standard AA |
|
M |
Met |
Flexible sulfur geometry; oxidizable to sulfoxide in long MD runs; check reactive FF |
Translation initiation; hydrophobic core; redox sensing |
Claude generated most of the above information. We’ll add onto the md_note section as we encounter certain things during our MD sims.
What’s Zwitterion?
A zwitterion is a molecule that has both positive and negative charges but is overall electrically neutral. In amino acids, the amino group (–NH₂) can accept a proton to become positively charged (–NH₃⁺), while the carboxyl group (–COOH) can lose a proton to become negatively charged (–COO⁻). At physiological pH (~7.4), most amino acids exist as zwitterions, with the amino group protonated and the carboxyl group deprotonated. This dual charge allows amino acids to interact with both polar and non-polar environments, contributing to their solubility in water and their ability to form various interactions in proteins.
What Does Non-polar Actually Mean?
It is worth clarifying what “non-polar” actually refers exclusively to the side chain (R group) — specifically that it consists largely of carbon and hydrogen bonds with no net dipole and no ionizable groups, making it hydrophobic and largely indifferent to water. It says nothing about the backbone, which is the same for all amino acids and always carries polar bonds (C=O, N–H). In fact, as mentioned above, all amino acids including non-polar ones exist as zwitterions at physiological pH — a property that comes entirely from the backbone, not the side chain.
Note to self: All amino acids’ backbones are zwitterions; the R-side chain determines polarity and hydrophobicity. Also, net charge neutral == overall charges equals zero, does not mean the molecule is non-polar.
Rodriguez Rotation Formula
Rodrigues’ rotation formula is a method for rotating a 3D vector in space around a specified axis by a given angle. The formula is expressed as:
\(v_{rotation} = v.\cos(\theta) + \sin(\theta)(k \times v) + (1 - \cos(\theta))(k(k \cdot v))\)
where v is the original vector, k is the unit vector along the axis of rotation, and θ is the angle of rotation in radians.

This formula apparently is very popular in computer graphics and robotics, but I can see how it can be useful in molecular dynamics as well when we want to rotate a molecule or a part of it around an axis. Especially when we want to estimate the least energy conformation of a molecule. The direction application of this formula in amino acid sequence would be in rearranging the atoms based on phi and psi which are the angles of rotations around the N-Cα and Cα-C bonds of the amino acid backbone, respectively. By applying Rodrigues’ rotation formula, we can calculate the new positions of the atoms in the amino acid after rotating them by the specified angles, allowing us to explore different conformations of the molecule. How I remember which angle is which is Nancy Phi (sounds like some detective show and also N->C) and C C Psi (All with S sound, also Carbon to carbon). We’ll leave the hand calculation until next time, but let’s learn how to rotate a coordinate based on an axis with Rodriguez!
Below I’ll write the code first, then explain. Please feel free to use your mouse to hover over the plotly object and check out the coordinates.
library(plotly)
library(pracma)
#### Let's start simple
x1 <- c(0,0,0)
x2 <- c(1,1,1)
x3 <- c(1,2,1)
rodrigues <- function(v, k, theta) {
k <- k / sqrt(sum(k^2))
cos(theta)*v + sin(theta)*pracma::cross(k, v) + (1 - cos(theta))*sum(k*v)*k
}
k <- x2 - x1
v <- x3 - x1
result <- rodrigues(v,k,pi/2) #notice this, pi/2 == 90 degrees
pts <- data.frame(
x = c(x1[1],x2[1],x3[1]),
y = c(x1[2],x2[2],x3[2]),
z = c(x1[3],x2[3],x3[3]),
label = c("x1","x2","x3")
)
pts_rs <- data.frame(
x = c(x1[1],x2[1],result[1]),
y = c(x1[2],x2[2],result[2]),
z = c(x1[3],x2[3],result[3]),
label = c("x1","x2","x3_new")
)
plot_ly() |>
add_trace(data=pts, x=~x, y=~y, z=~z,
type="scatter3d", mode="lines+markers+text",
text=~label,
marker=list(size=8, color="blue", opacity=0.5),
line=list(width=4, color="blue", dash="solid")) |>
add_trace(data=pts_rs, x=~x, y=~y, z=~z,
type="scatter3d", mode="lines+markers+text",
text=~label,
marker=list(size=8, color="red", opacity=0.5),
line=list(width=4, color="red", dash="dash"))
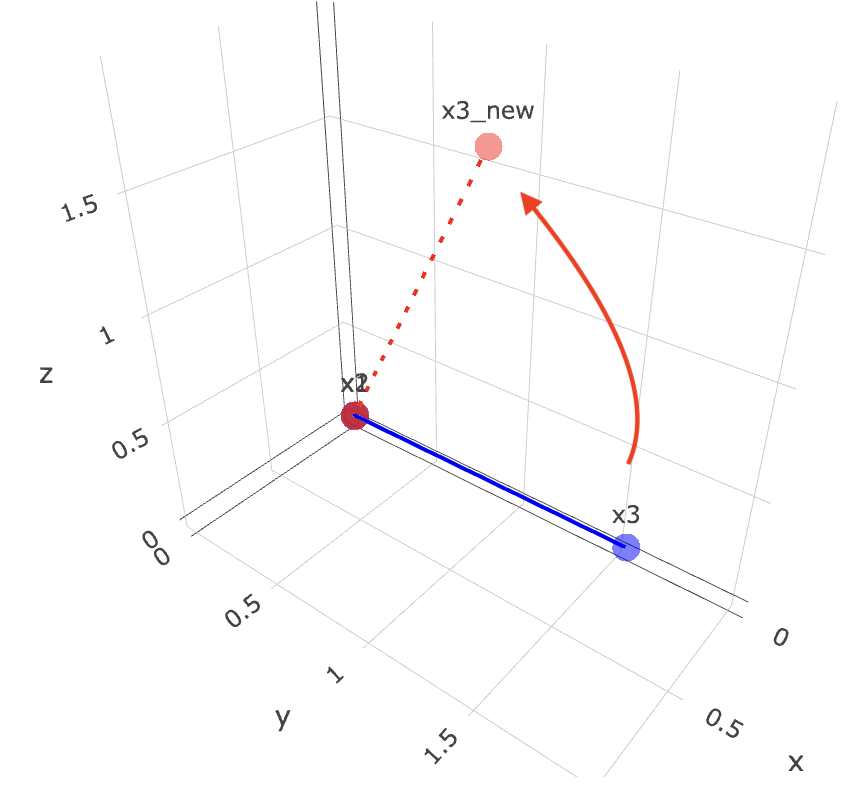
So with the above, we want to start off with 3 points, x1, x2, x3. they all represent their xyz coordinates.
Funny thing is, xyz coordinate here is different from what I learnt xyz as. I’ve always thought x is horizontal, y is vertical and z is depth. But in this case, x is depth, y is horizontal and z is vertical. I guess it depends on how you look at it.
Then we want to rotate x3 around the axis defined by x1 and x2 by 90 degrees (pi/2 radians). The rodrigues function takes in the vector v (which is the vector from x1 to x3), the axis k (which is the vector from x1 to x2), and the angle theta (which is pi/2). It returns the new coordinates of x3 after rotation.
Finally, we plot the original points and the rotated point using plotly. The original points are in blue, and the rotated point is in red. You can hover over the points to see their coordinates.
Now if we were to maneuver the 3d plot and align both x1 and x2 into a dot, we can clearly see that it moved 90 degrees anti-clockwise!
Now there is a pretty cool rule to know where the rotation should occur anti-clockwise vs clockwise is by using your hand !!! Remember this from high school?
It would be really cool to derive the above formula. There are a lot of videos that have done this. I’m still trying to conceptualize it, let’s leave that for another blog! It sounds interesting and may be a good exercise, especially when we’re venturing into 3d spaces.
Lennard-Jones Potential Energy
The Lennard-Jones potential energy formula is a mathematical model used to describe the interaction between a pair of neutral atoms or molecules. It is given by the equation:
\(V(r) = 4\epsilon \left[ \left( \frac{\sigma}{r} \right)^{12} - \left( \frac{\sigma}{r} \right)^6 \right]\)
Where:
\(V(r)\)is the potential energy as a function of the distance\(r\)between the two particles.\(\epsilon\)is the depth of the potential well, representing the strength of the attractive interaction.\(\sigma\)is the finite distance at which the inter-particle potential is zero, representing the effective diameter of the particles.- The term
\(\left( \frac{\sigma}{r} \right)^{12}\)represents the repulsive part of the potential, which dominates at short distances due to the Pauli exclusion principle. - The term
\(\left( \frac{\sigma}{r} \right)^6\)represents the attractive part of the potential, which dominates at longer distances due to van der Waals forces.
The Lennard-Jones potential is widely used in molecular dynamics simulations to model the interactions between non-bonded atoms or molecules, particularly in the context of van der Waals forces. It helps to predict the behavior of particles in a system, such as their equilibrium positions and the energy landscape of molecular interactions.
Wow there are a bunch of terms and word above! I’m getting dizzy just to keep track of what is what. Let’s push through this. To use the above formula, we’d have to have some understanding of the parameters epsilon and sigma. These parameters are typically derived from experimental data or quantum mechanical calculations and are specific to the types of atoms or molecules involved in the interaction. Where to get these parameters?
Here you go - openbabel:: gaff.dat
When you opened gaff.dat there are bunch of numbers! Let’s find the numbers that are meaningful for us. All the below are separated by new lines as you scroll down.
Bond Stretch
The column names should be: type mass(g/mol) polarizability(ų) source

Bond Angle
The column names should be: types K r0 source count rmsd
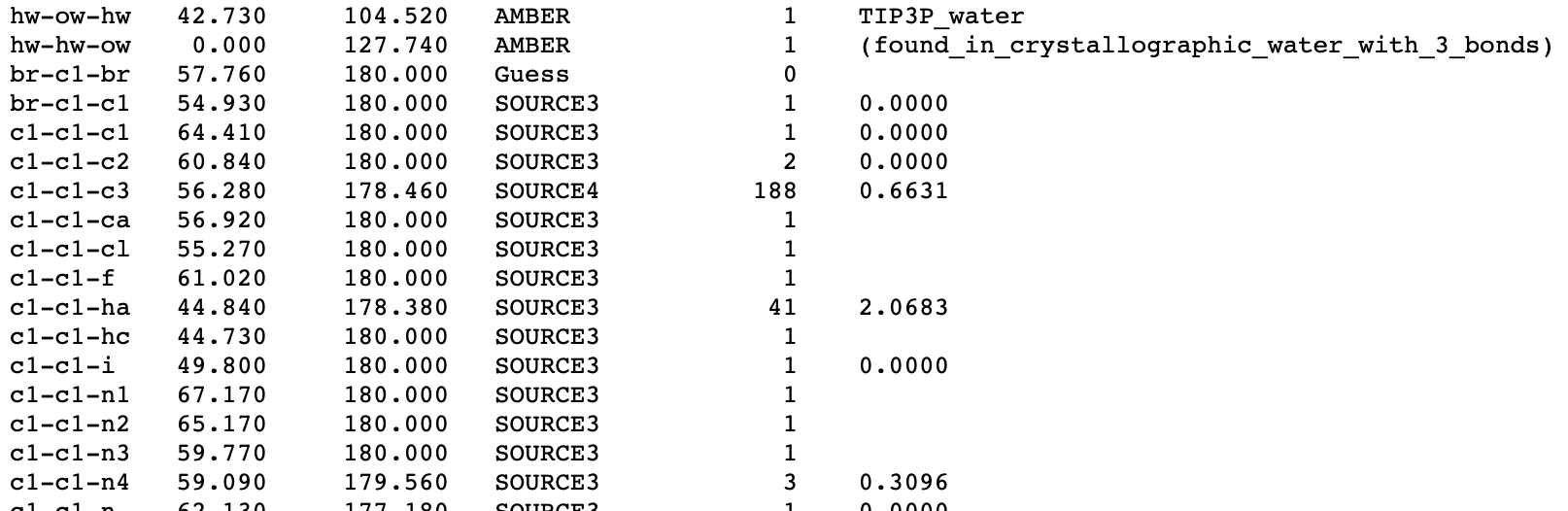
Proper Dihedral
The column names should be: types div barrier phase periodicity

Non-bonded
The column names should be: type R*(Å) ε(kcal/mol)

We are purely interested in the non-bonded section where R* is our sigma = R* × 2 / 2^(1/6) and ε is our epsilon. With the above parameters, we can then calculate the Lennard-Jones potential energy between any two atoms in a molecule. Let’s do a simple calculation for ethanol.
Calculating LJ
library(tidyverse)
library(igraph)
# Coordinates (x, y, z) in Angstroms, according to pubchem ethanol molecule
coords <- rbind(
O = c( -1.1712, 0.2997, 0.0000),
C2 = c( -0.0463, -0.5665, 0.0000),
C1 = c( 1.2175, 0.2668, 0.0000),
H4 = c( -0.0958, -1.2120, 0.8819),
H5 = c( -0.0952, -1.1938, -0.8946),
H1 = c( 2.1050, -0.3720, -0.0177),
H2 = c( 1.2426, 0.9307, -0.8704),
H3 = c( 1.2616, 0.9052, 0.8886),
H6 = c( -1.1291, 0.8364, 0.8099)
)
# AMBER GAFF parameters (sigma Å, epsilon kcal/mol)
Rstar_to_sigma <- function(Rstar) 2 * Rstar / 2^(1/6)
sigma <- c(
C1=Rstar_to_sigma(1.9080), C2=Rstar_to_sigma(1.9080),
O=Rstar_to_sigma(1.7210),
H1=Rstar_to_sigma(1.4870), H2=Rstar_to_sigma(1.4870),
H3=Rstar_to_sigma(1.4870), H4=Rstar_to_sigma(1.4870),
H5=Rstar_to_sigma(1.4870), H6=0.0000
)
epsilon <- c(
C1=0.1094, C2=0.1094, O=0.2104,
H1=0.0157, H2=0.0157, H3=0.0157,
H4=0.0157, H5=0.0157, H6=0.0000
)
# Bonds
bonds <- tribble(
~from, ~to,
"C1", "C2",
"C2", "O",
"O", "H6",
"C1", "H1",
"C1", "H2",
"C1", "H3",
"C2", "H4",
"C2", "H5"
)
# count bonds between two atoms
g <- graph_from_data_frame(bonds, directed = FALSE)
g_dist <- distances(g)
# LJ function
lj <- function(r, eps, sig) 4 * eps * ((sig/r)^12 - (sig/r)^6)
# Loop all pairs
atoms <- rownames(coords)
pairs <- combn(atoms, 2, simplify=FALSE) # combn so we don't repeat
total_V <- vector(mode = "numeric", length = length(pairs))
for (i in 1:length(pairs)) {
# each pair
p <- pairs[[i]]
from <- p[1]
to <- p[2]
num_bond <- g_dist[from, to]
if (num_bond <= 2) next
# params needed for LJ
r <- sqrt(sum((coords[from,] - coords[to,])^2))
sig <- (sigma[from] + sigma[to]) / 2
eps <- sqrt(epsilon[from] * epsilon[to])
# scale if num bond is 3 (4 atoms)
scale <- if (num_bond == 3) 0.5 else 1.0
# LJ calc
V <- scale * lj(r, eps, sig)
cat(from, "-", to, " num of bonds=", num_bond, " r=", r, " V=", V, "\n")
total_V[i] <- V
}
## O - H1 num of bonds= 3 r= 3.344395 V= -0.02733269
## O - H2 num of bonds= 3 r= 2.642383 V= 0.110613
## O - H3 num of bonds= 3 r= 2.659841 V= 0.09535471
## C1 - H6 num of bonds= 3 r= 2.546942 V= 0
## H4 - H1 num of bonds= 3 r= 2.521587 V= 0.01461147
## H4 - H2 num of bonds= 3 r= 3.074579 V= -0.007593085
## H4 - H3 num of bonds= 3 r= 2.514978 V= 0.01576022
## H4 - H6 num of bonds= 3 r= 2.295394 V= 0
## H5 - H1 num of bonds= 3 r= 2.507028 V= 0.01720963
## H5 - H2 num of bonds= 3 r= 2.510736 V= 0.01652426
## H5 - H3 num of bonds= 3 r= 3.070262 V= -0.007612401
## H5 - H6 num of bonds= 3 r= 2.845344 V= 0
## H1 - H6 num of bonds= 4 r= 3.550289 V= 0
## H2 - H6 num of bonds= 4 r= 2.908137 V= 0
## H3 - H6 num of bonds= 4 r= 2.392984 V= 0
cat("\nTotal V_LJ:", sum(total_V), "kcal/mol\n")
##
## Total V_LJ: 0.2275351 kcal/mol
Alright, the above we basically were trying to calculate all pairwise atoms that is more than 3 bonds (if it’s exactly 3 bonds, we scale it by half). Alright, now that we know how to do that on simple molecular, next time we can use this to minimize on as we’re seeking optimal phi and psi!
Opportunities For Improvement
- Derive Rodriguez Rotation formula
- put both Rodriguez rotation formula and LJ calculation into action to find the optimal phi and psi of amino acid sequence of a protein!
- need to include secondary/tertiary structure interactions too
Lessons learnt
- refreshed on rotation and vectors
- learnt rodriguez rotation formula
- learnt LJ formula
- learnt what gaff.dat actually contains.
- learnt about phi and psi and what they actually mean.
- learnt about net charge == total charge; polar vs non-polar is R-chain dependent.
If you like this article:
- please feel free to send me a comment or visit my other blogs
- please feel free to follow me on BlueSky, twitter, GitHub or Mastodon
- if you would like collaborate please feel free to contact me
- Posted on:
- June 7, 2026
- Length:
- 13 minute read, 2722 words